PGATK NF-Core Workflows¶
The ProteoGenomics Analysis Toolkit provides a set of workflows to perform large scale proteogenomics data analysis. All workflows are developed using nextflow and BioContainers following nf-core.
All PGATK workflows are deposited in nf-core.
Requirements¶
Starting with Nextflow¶
Nextflow can be used on any POSIX compatible system (Linux, OS X, etc). It requires Bash 3.2 (or later) and Java 8 (or later, up to 11) to be installed.
Installation, it only needs two easy steps:
Download the executable package by copying and pasting the following command in your terminal window:
1 | wget -qO- https://get.nextflow.io | bash
|
You can read more about how to setup your nf-core or nextflow enviroments.
PGDB: proteogenomics database generation¶
The ProteoGenomics DataBase (pgdb) generation pipeline is a nf-core workflow that enables the generation of custom proteogenomics databases for MS proteomics studies using the pypgatk library.
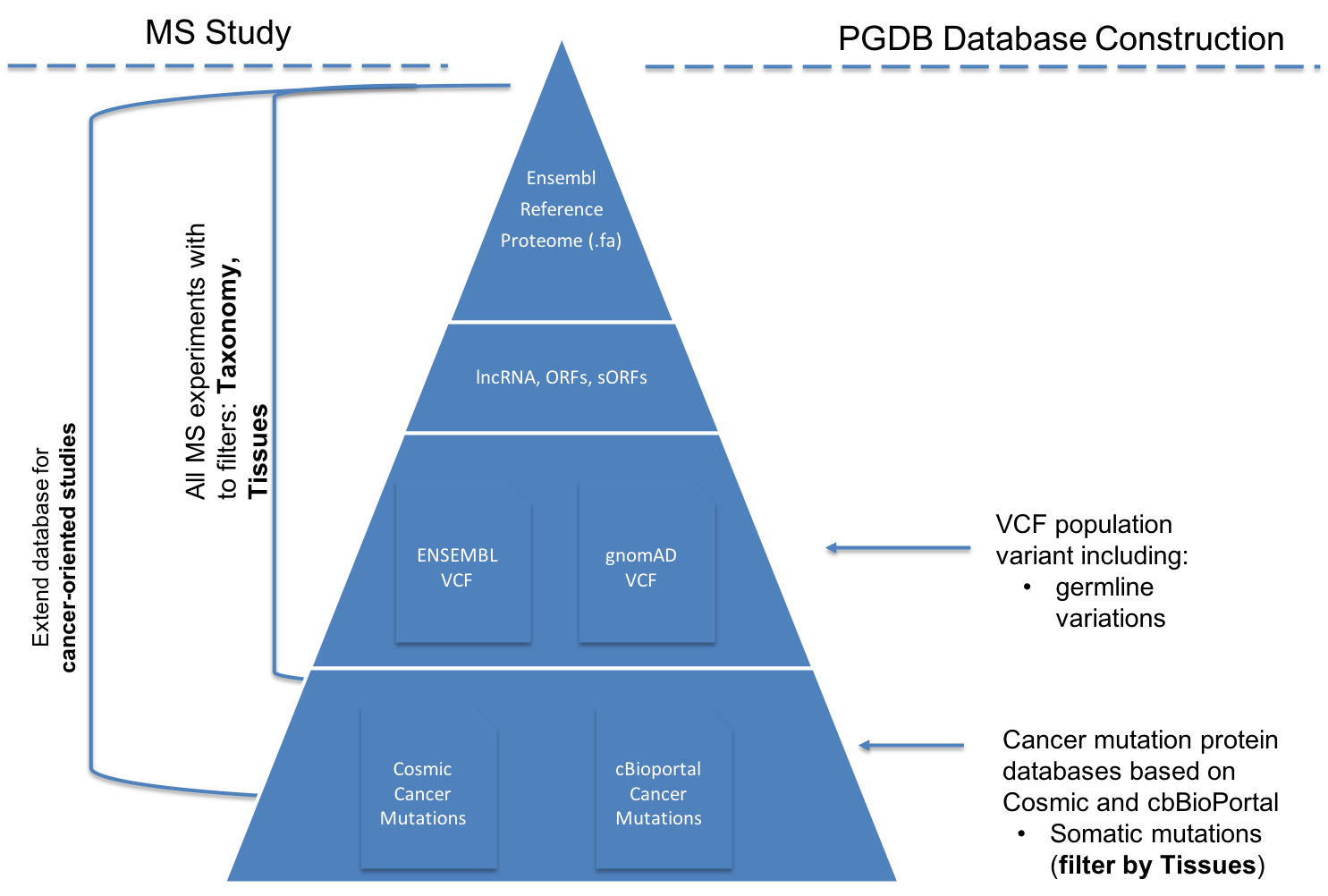
The pgdb pipelines enable to generate a variety of ENSEMBL-based proteognomics databases depending of the type of study.
Workflow usage¶
All workflow options can be seen by using the --help command:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 | N E X T F L O W ~ version 19.04.1
Launching `main.nf` [sleepy_stonebraker] - revision: 9cff592eaf
Usage:
The typical command for running the pipeline is as follows:
nextflow run main.nf --taxonomy 9606 --ensembl false --gnomad false --cosmic false --cbioportal false
This command will generate a protein datbase for non-coding RNAs, pseudogenes,
altORFs. Note the other flags are set to false.
A final fasta file is created by merging them all and the canonical
proteins are appended. The resulting database is stored in result/final_proteinDB.fa
and its decoy is stored under result/decoy_final_proteinDB.fa
Options:
Process flags
--ncrna [true | false] Generate protein database from non-coding RNAs
--pseudogenes [true | false] Generate protein database from pseudogenes
--altorfs [true | false] Generate alternative ORFs from canonical proteins
--cbioportal [true | false] Download cBioPortal studies and genrate protein database
--cosmic [true | false] Download COSMIC files and generate protein database
--ensembl [true | false] Download ENSEMBL variants and generate protein database
--gnomad [true | false] Download gnomAD files and generate protein database
--decoy [true | false] Append the decoy proteins to the database
Configuration files By default all config files are located in the configs
directory.
--ensembl_downloader_config Path to configuration file for ENSEMBL download parameters
--ensembl_config Path to configuration file for parameters in generating
protein databases from ENSMEBL sequences
--cosmic_config Path to configuration file for parameters in generating
protein databases from COSMIC mutations
--cbioportal_config Path to configuration file for parameters in generating
protein databases from cBioPortal mutations
--protein_decoy_config Path to configuration file for parameters used in generating
decoy databases
Database parameters:
--taxonomy Taxonomy (Taxon ID) for the species to download ENSEMBL data,
default is 9606 for humans.
For the list of supported taxonomies see:
https://www.ensembl.org/info/about/species.html
--cosmic_tissue_type Specify a tissue type to limit the COSMIC mutations to
a particular caner type (by default all tumor types are used)
--cbioportal_tissue_type Specify a tissue type to limit the cBioPortal mutations to
a particular caner type (by default all tumor types are used)
--af_field Allele frequency identifier string in VCF Info column,
if no AF info is given set it to empty.
For human VCF files from ENSEMBL the default is set to MAF
Output parameters:
--final_database_protein Output file name for the final database protein fasta file
under the result/ directory.
--decoy_prefix String to be used as prefix for the generated decoy sequences
--result_file Output file-path for the final database, not under the result folder.
Data download parameters:
--cosmic_user_name User name (or email) for COSMIC account
--cosmic_password Password for COSMIC account
In order to be able to download COSMIC data, the user should
provide a user and password. Please first register in COSMIC
database (https://cancer.sanger.ac.uk/cosmic/register).
--gencode_url URL for downloading GENCODE datafiles:
gencode.v19.pc_transcripts.fa.gz and
gencode.v19.annotation.gtf.gz
--gnomad_file_url URL for downloading gnomAD VCF file(s)
--help Print this help document
========================================================================================
Pipeline Tasks:
========================================================================================
Get fasta proteins, cdnas, ncRNAs and gtf files from ENSEMBL (default species = 9606)
(processes: ensembl_fasta_download, gunzip_ensembl_files, merge_cdnas)
Generate ncRNA, psudogenes, altORFs databases
(processes: add_ncrna, add_pseudogenes , add_altorfs)
Generate ENSEMBL variant protein database (VCFs, default species = 9606)
(processes: ensembl_vcf_download, gunzip_vcf_ensembl_files, check_ensembl_vcf, ensembl_vcf_proteinDB)
Generate gnomAD variant protein database
(processes: gencode_download, , extract_gnomad_vcf, gnomad_proteindb)
Generate COSMIC mutated protein database (default all cancer types)
(processes: cosmic_download , gunzip_cosmic_files, cosmic_proteindb)
Generate cBioPortal mutated protein database (default all studies and all cancer types)
(processes: cds_GRCh37_download, download_all_cbioportal, cbioportal_proteindb)
Concatenate all generated databases
(processes: merge_proteindbs)
Generate a decoy database from the concatenated database
(processes: decoy)
----------------------------------------------------------------------------------------
|
Seudo-genes, long non-coding RNAs¶
If the study attempt to identified novel pseudo-genes, long non-coding RNA peptides and proteins in Human, the users can generate the database by concatenating the ENSEMBL Human reference proteome and the novel coding regions using the following command:
1 | nextflow run main.nf --taxonomy 9606 --ensembl false --gnomad false --cosmic false --cbioportal false --altorfs false -profile local,standard -c nextflow.config
|
This command will attached to the reference ENSEMBL Human proteome, the pseudo-genes and (pipeline option –pseudogenes) and the long non-coding RNA peptides (–ncrna).
Note
Most of the options in the pipeline are enable by default. For example –add_reference, includes in the results database the reference proteome for the species under study.
COSMIC and cBioPortal Variants¶
If COSMIC variants wants to be added to the database, the following command can be used:
1 | nextflow run main.nf --taxonomy 9606 --ensembl false --gnomad false --cosmic true --cbioportal false --altorfs false --cosmic_user_name username --cosmic_password password -profile local,standard -c nextflow.config
|
Note
For COSMIC database a user and password should be provided to the pipeline to be able to download the database variants and the celllines information.